



造纸植物纤维原料和纸浆中的糖类包括纤维素和半纤维素。其中,纤维素是由单一的葡萄糖基组成的,酸水解产物为单一的葡萄糖;而半纤维素则由多种糖基组成,酸水解后的主要单糖有葡萄糖、甘露糖、木糖、阿拉伯糖和半乳糖等。
各种原料中糖类的组成和结构不同,而且各种糖类组分在制浆造纸过程中的溶出情况是不相同的。因此,除需要测定原料和纸浆中纤维素和半纤维素的总含量外,再进一步分析其中各种碳水化合物组分的各自含量,对于表征原料的特性和研究制浆造纸过程中各糖类组分的溶出规律具有重要意义。
造纸原料和纸浆中糖类组成的分析通常采用色谱法(包括纸色谱法、薄层色谱法、气相色谱法和液相色谱法)。气相色谱法自1963年开始用于造纸原料和纸浆的糖类组分分析,由于其具有分离效能高,分析速度快,样品用量少,定量结果准确,易于自动化等优点,因此成为造纸现代测试技术中比较成熟的,且应用较广泛的测定方法之一。气相色谱-质谱联用(GC-MS)也成功地用于糖类组分的分析。
采用气相色谱法分析造纸植物纤维原料和纸浆中糖类的相对组成,既分别求得糖类中各主要单糖(葡萄糖、甘露糖、木糖、半乳糖、阿拉伯糖等)的相对百分数(相对含量),还可计算出各种单糖对原料(或纸浆)绝干量的百分数(绝对含量)。
下面对气相色谱法糖类组分析的操作程序做一简述,并介绍造纸原料和纸浆中糖类组分气相色谱分析的国家标准方法(《GB/T 12033—2008造纸原料和纸浆中糖类组分的气相色谱的测定》)。同时,国外文献介绍了对糖类组分气相色谱分析方法的一些改进措施,本节也略加节选,供大家多考,以便于简化操作。
(一)气相色谱分析的程序
气相色谱法糖类组分的分析,需首先制备糖类挥发性衍生物,以适应气相色谱对分析试样的要求;要选择合适的担体和固定液种类;还要测定出各种单糖的重量校正因子;色谱分析后依据气相色谱的相对保留时间进行定性分析,以确定各色谱峰所代表的单糖种类;继而进行定量分析,最终计算出各种单糖的含量(相对含量和绝对含量)。下面对上述程序做一简述:
1.糖类挥发性衍生物的制备方法与原理
气相色谱分析对试样的要求是能气化,而且热稳定性好,不分解。因此,造纸植物纤维原料和纸浆试样采用气相色谱法分析糖类组成时,必须首先对试样进行处理,先用硫酸使聚糖通过酸水解而生成醛糖,然后再制备成单糖的挥发性衍生物。常用的制备糖类挥发性衍生物的方法有:三甲基硅醚化法、糖腈乙酸酯化法和糖醇乙酸酯化法等。我国已将造纸原料和纸浆中糖类组分的糖醇乙酸酯化法气相色谱分析定为国家标准测定方法(GB/T 12033—2008)。
(1)糖三甲基硅醚化法
将酸水解后生成的醛糖溶解于吡啶中,加入硅醚化试剂(六甲基二硅胺和三甲基氯硅烷)进行硅醚化,生成各种糖的三甲基硅醚衍生物。反应式如下:
该方法制备较简单,但三甲基硅醚化糖衍生物存在异构体,在色谱图上出现一种单糖有多个色谱峰,或是几种单糖峰互有重叠的情况,给定量带来困难,因而现在较少采用。
(2)糖腈乙酸酯化法
将醛糖溶解于吡啶后,加入盐酸羟胺,使醛糖转变成糖肟,再与乙酸酐反应生成糖腈乙酸酯。反应式如下:
该法较为简单,无异构体,定量较准确。
(3)糖醇乙酸酯化法
醛糖用硼氢化钠(NaBH4)还原成糖醇以后,用乙酸酐和乙酸丁酯将糖醇乙酰化,即转变为糖醇乙酸酯。反应式如下:
该法测定精确,但制样较复杂。GB/T 12033—2008采用糖醇乙酸酯化法。
2.担体和固定相的选择
气相色谱法分析糖类组分常选用的担体和固定液种类如下:
担体:Chromosorb W A W DMCS;Gas-chrom QP(一种经酸洗、碱洗和硅烷化处理的硅藻土型白色担体)。
固体液:3%ECNSS-M(丁二酸乙二醇-氰乙基硅氧烷的共聚物);OV-17、OV-225(硅氧烷);XF-1112;1.5%EGS+1.5XE-60(乙二酸乙二醇酯和氰乙基甲基硅酮)。
GB/T 12033—2008采用Chromosorb W A W DMCS作为担体,固定液为3%ECNSS-M。
3.各种单糖质量校正因子的求法
某一组分的质量与色谱峰面积之比,称为该组分的绝对质量校正因子。在定量分析中都是用相对质量校正因子,即某组分与一种标准物质(内标物)的绝对校正因子之比值。糖类组分测定的内标物为肌醇。
某一组分的相对质量校正因子Ki按式(8-31)计算:
式中 mi——某一组分质量,mg
ms——内标物的质量,mg
Ai——某一组分的色谱峰面积
As——内标物的色谱峰面积
4.定性分析
将待测试样(原料或纸浆)和内标物(肌醇)先制备成糖的挥发性衍生物,然后进行气相色谱分析,得到试样的色谱图(如图8-7为鱼鳞松气相色谱图),图中各种单糖组分已得到分离。
定性分析即是确定待测试样色谱图中各个色谱峰所对应的单糖的种类。通常依据相对保留时间来确定。
就一个色谱峰而言:被测组分从进样到流出的最大浓度(色谱峰的峰值)所经过的时间称为保留时间(tR);由保留时间扣除空气峰的出峰时间(死时间t0)的差值,称为校正保留时间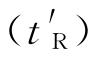 ;相对保留时间则为被测组分的校正保留时间
;相对保留时间则为被测组分的校正保留时间 与内标物的校正保留时间
与内标物的校正保留时间 之比值。即:
之比值。即:
图8-7 鱼鳞松的糖醇乙酸酯气相色谱图
(色谱柱Gas Chrom Q涂 3%ECNSS—M)
1—溶剂峰 2—空气峰 3—阿拉伯糖 4—木糖 5—甘露糖 6—半乳糖 7—葡萄糖 8—肌醇(内标物)
取5种纯的标准单糖(葡萄糖、甘露糖、木糖、半乳糖和阿拉伯糖)试剂,加入内标物(肌醇),按与试样相同的条件,分别制备糖的挥发性衍生物和进行色谱分析,作出各标准单糖的气相色谱图。
在一定的色谱条件下,各组分有一定的保留时间,不受混合物中共存的其他组分的影响。因此,在同一色谱条件下,将得到的试样色谱图与已知标准单糖的色谱图相对照,根据相对保留时间可确定被测定试样色谱图中的某一色谱峰是由何种单糖所产生。
5.定量分析
气相色谱的定量分析方法主要有下列3种:
(1)归一化法
当试样中的所有组分都能产生相应的色谱峰,并且已知各个组分的相对质量校正因子,则可用归一化法求出各组分的含量:
式中 wi——组分i的百分含量,%
A1、A2…An——组分1、2……n的色谱峰面积
K1、K2…Kn——组分1、2……n的重量校正因子
(2)内标法
内标物的色谱峰应当是与试样中各组分的色谱峰互不重叠,且出峰时间相近。测定造纸原料和纸浆中糖类组分时,通常用肌醇作为内标物。
试样中各组分的百分含量wi用式(8-34)计算:
式中 m——试样质量,mg
mi——某一组分质量,mg
ms——内标物质量,mg
Ai——某一组分的色谱峰面积
Ki——某一组分的相对重量校正因子
As——内标的色谱峰面积
内标法准确度高,操作比较麻烦。
(3)外标法
又称为已知样校正法。即首先用纯的标准试剂作出标准曲线(峰面积与含量的关系曲线)。在同样的色谱条件下,并且准确而定量地进样(即进样量相等),则根据色谱面积,可由标准曲线查得某一组分的含量。
气相色谱法分析造纸植物纤维原料和纸浆中糖类组分时,通常采用内标法(以肌醇作内标物),也有归一化法的。
(二)造纸原料和纸浆中糖类组分的气相色谱测定方法
参见国家标准GB/T 12033—2008。
本标准规定了造纸原料和纸浆中主要糖类组分的气相色谱测定方法。本标准适用于各种造纸原料及各种纸浆中糖类组分的测定。
1.测定原理
用硫酸在高温高压的条件下,将造纸原料或纸浆中的纤维素和半纤维素水解成单糖溶液,以碳酸铅中和后采用硼氢化钠进行还原,使之成为糖醇。在高温条件下,用乙酸酐进行衍生化,形成挥发性衍生物,然后进行气相色谱分析,内标法定量。
2.试剂和材料
除非另有说明,在分析中仅使用确认为分析纯的试剂和蒸馏水或去离子水或相当纯度的水。
a.碱 式 碳 酸 铅[Pb(OH)2· 2PbCO3];b.硼 氢 化 钠(NaBH4);c.乙 酸(CH3COOH):浓度为36%;d.二氯甲烷(CH2Cl2):色谱纯;e.无水乙醇(C2H5OH);f.乙酸酐(C4H6O3);g.乙酸丁酯(C6H12O2);h.硫酸溶液:用蒸馏水将优级纯的硫酸配制成浓度为72%;i.盐酸溶液:浓度为1mol/L;j.内标物:肌醇标准品(C6H12O6),纯度大于98.5%;k.葡萄糖标准品(C6H12O6):纯度大于98.5%;l.半乳糖标准品(C6H12O6):纯度大于98.5%;m.甘露糖标准品(C6H12O6):纯度大于98.5%;n.木糖标准品(C5H10O5):纯度大于98.5%;o.阿拉伯糖标准品(C5H10O5):纯度大于98.5%;p.内标物溶液:准确称取适量内标物,用蒸馏水配制成所需浓度的内标物溶液;q.混合标准溶液:准确称取适量的葡萄糖标准品、半乳糖标准品、甘露糖标准品、木糖标准品、阿拉伯糖标准品及内标物,用蒸馏水配制成所需浓度的混合标准溶液;r.强酸型阳离子交换树脂。
3.仪器和设备(https://www.xing528.com)
实验室常用仪器及以下仪器。
a.气相色谱仪:配有氢火焰检测器(FID)。b.电子天平,感量0.0001g。c.恒温水浴锅。d.医用高压锅。e.旋转蒸发器:可控制温度至75℃,能抽真空至-101kPa(-750mmHg)。f.烘箱:可调至(120±2)℃。g.真空干燥器,干燥剂为五氧化二磷。
4.试样制备
如果是原料测定,应将原料磨碎至40~60目;如果是纸浆测定,应将纸浆抄成纸片后撕碎。风干24h后,各称取2份2g试样,按GB/T 462—2008测定其绝干质量。
5.试验步骤
(1)水解
称取绝干质量约为0.3g(准确至0.0005g)的试样于15mL离心管中,置于真空干燥器内,抽真空后,放置过夜。移取3mL硫酸溶液加入其中,插入尖头玻璃棒搅拌均匀,在(30±1)℃水浴锅中水浴1h进行水解,用66mL蒸馏水分数次清洗至200mL烧杯中。加入10mL内标物溶液后,置于医用高压锅中,在120℃水解1h,取出。
(2)还原
上述水解溶液冷却后,从中取10mL清液于50mL烧杯中,加适量碱式碳酸铅,中和pH至5,过滤。在滤液中加入0.2g硼氢化钠,置于(40±1)℃水浴锅中水浴0.5h,然后滴加乙酸至无气泡为止。将此溶液转移至10mL量筒中,加蒸馏水至10mL刻度,用玻璃棒轻轻上下搅均后进行离子交换。
(3)离子交换
将强酸型阳离子交换树脂风干,磨碎至150~200目。用蒸馏水浸泡数小时后,注入内塞有少许玻璃棉的10mL酸式滴定管中约3mL,制备成强酸型阳离子交换柱,用盐酸溶液淋洗,使其成为H+型。然后用蒸馏水洗涤该强酸型阳离子交换柱内残余的盐酸,直至中性为止。移取1mL上述还原并稀释后的溶液,注入该强酸型阳离子交换柱,再用蒸馏水淋洗,流速控制约为0.2mL/min,直至流出液为中性。
(4)衍生化
将所收集到的流出液用旋转蒸发器在55℃浓缩至干,加人5mL无水乙醇,蒸干,再加入5mL无水乙醇,再蒸干。加入乙酸酐和乙酸丁酯1mL,在(120±2)℃的烘箱中酯化1.5h。取出后,加5mL蒸馏水,用旋转蒸发器在75℃浓缩至干,再加1mL蒸馏水,蒸干,用1mL二氯甲烷定容后,供气相色谱分析。
(5)标准工作液的制备
准确吸取适量的混合标准溶液,按(2)、(3)、(4)步骤操作。
(6)测定
1)气相色谱条件
由于测定结果取决于所使用的仪器,因此不可能给出气相色谱的通用参数。设定的参数应保证色谱测定时被测定组分与其他组分能够得到有效的分离,下面给出的参数证明是可行的。
① 色谱柱:30m×0.32mm(内径)×0.25μm(膜厚),DB-5 石英毛细管柱;
② 色谱柱温度程序:220℃保持15min,然后以2℃/min程序升温至260℃,保持10min;
③ 进样口温度:300℃;
④ 检测器温度:300℃;
⑤ 载气:氮气,纯度≥99.99%,0.7mL/min;
⑥ 进样方式:分流进样,分流比为1∶20,0.75min后开阀;
⑦ 进样量:1.0μL。
2)气相色谱测定
根据样液中被测物的含量情况,选定浓度相近的标准工作溶液。按1)的条件,分别对标准工作液和试样溶液进行分析。用色谱峰保留时间定性,内标法定量,标准工作液和样品溶液中的单糖衍生物的响应值应在仪器检测的线性范围内。单糖衍生物的保留时间参见附录A,典型气相色谱图参见附录B。
6.结果计算
按式(8-35)计算各单糖校正因子K:
式中 Ac——标准工作溶液中标准单糖色谱峰面积
mc——标准工作溶液中标准单糖的质量,mg
As——标准工作溶液中内标物色谱峰面积
ms——标准工作溶液中内标物质量,mg
注:确定K需称5种标准单糖,各种标准单糖称量数值应尽量接近样品中实际的各种单糖的质量。一般要求的分析按下述称量:葡萄糖0.13g,半乳糖和阿拉伯糖各0.01g,甘露糖和木糖各0.03g。
按式(8-36)和式(8-37)计算试样中单糖和聚糖的绝对含量(%):
式中 A——试样溶液中各单糖的峰面积
As——试样溶液中内标物峰面积
ms——试样溶液中内标物质量,mg
m——试样的绝干质量,g
B——单糖与聚糖的转换系数
对葡萄糖、半乳糖、甘露糖,B=0.9;对木糖、阿拉伯糖,B=0.88。
按式(8-38)的和式(8-39)计算试样中单糖和聚糖的相对含量(%):
7.检测低限和回收率
(1)检测低限
本标准对阿拉伯糖、木糖、葡萄糖、甘露糖、半乳糖的检测低限均为0.1%。
(2)回收率
本标准中5种糖类组分的回收率见表8-10。
表8-10 5种糖类组分的回收率
(3)精密度
在同一实验室,由同一操作者使用相同设备,按相同的测定方法,并在短时间内对同一被测对象相互独立进行测定,所获得的两次独立测定结果的绝对差值不大于这两个测定值的算术平均值的10%。以大于这两个测定值的算术平均值的10%的情况不超过5%为前提。
附录A(资料性附录)
5种单糖及内标物的衍生物气相色谱保留时间
5种单糖及内标物的衍生物气相色谱保留时间,见表8-11。
表8-11 5种单糖及内标物的衍生物气相色谱保留时间
附录B(资料性附录)
5种单糖及内标物衍生物的典型气相色谱图
图8-8给出了5种单糖及内标物衍生物的典型气相色谱图。
图8-8 5种单糖及内标物衍生物的典型气相色谱图
1—阿拉伯糖[L(+)arabinose] 2—木糖[D(+)xylose] 3—肌醇(inositol)4—无水葡萄糖(glucose anhydrous)5—甘露糖(D-mannose)6—半乳糖[D(+)galactose]
(三)气相色谱法测定糖类组分的改进方法
采用糖醇乙酸酯化气相色谱法分析糖类组分,虽然具有精确度高、定量准确的优点,但由于在糖醇乙酸酯制备过程中所需蒸发步骤较多,因而相当耗时费力。为了简化操作,改进分析效率,Tappi Journal1997Vol80(9)介绍了快速改良的气相色谱法用于纸浆碳水化合物组分分析的方法。
这种快速改良的气相色谱法是在醛糖的乙酰化过程中使用1-甲基咪唑作为催化剂。在这种催化剂的作用下,在分析过程中就不需要重复进行蒸发步骤,从而实现了快速、完全的糖醇乙酰化。经过对多种纸浆采用改良的方法和常规TAPPI测试方法T249cm-85所得测定结果的对比,表明该改良方法具有好的准确性和重复性。现将这种改良方法介绍给读者供参考。
操作步骤如下:取大约5g纸浆作样品,研磨过40目筛。同时使样品达到水分平衡。称取2份(0.35±0.01)g试样(精确到0.1mg)于150mL烧杯中,用移液管加入3mL 72%硫酸于试样中。用玻璃棒搅拌烧杯中的混合物,至试样开始溶解时为止。将烧杯置于(30±0.5)℃的水浴1h。每隔5~10min摇动一次。1h后加入84mL蒸馏水。用表面皿盖住烧杯,然后置于高压锅中,在121℃下加热1h。
加10mL内标物肌醇于烧杯中,混合后,将试样液冷却,木素残余物通过40mL多孔玻璃滤器坩埚过滤除去,保持一定真空度。最终的滤液调至140mL。加入大约11mL氢氧化铵溶液(含28.3%氨)于滤器中,使其中含1mol/L的NH3。移取2mL试样于125mL烧瓶中,称(35±1)mg硼氢化钠加入烧瓶中,将烧瓶置于(40±1)℃水浴中90min。其间摇动几次。结束时立即加入一滴冰醋酸抽提、分解过量的硼氢化钠,然后加入2mL1-甲基咪唑并同时搅拌,接着立即加入20mL醋酸酐于还原的单糖中。在室温下用瓷棒连续搅拌20min。所产生的热量加速了乙酰化反应的完成。
20min后,向烧瓶中添加30g碎冰和70g蒸馏水。继续搅拌至少20min。将混合物经一个250mL分液漏斗,用10、5、5mL二氯甲烷连续萃取,然后将二氯甲烷萃取物置于250mL烧瓶中。将烧瓶置于良好通风的排风罩中至少1h。将2mL二氯甲烷与干的醛糖乙酸混合。在20℃下将溶液贮存于4mL玻璃瓶中。在气相色谱前,将0.2mL醛糖乙酸溶液稀释于1mL二氯甲烷中,依据常规用注射器注入1μL稀释的醛糖乙酸溶液,判断5种单糖和内标物的峰面积,每一组分的相对百分含量根据TAPPI T249cm-85所给公式计算(参见国标GB/T 12033—2008计算方法和相对质量校正因子的测定方法)。
使用改良的方法有下列好处:在酸水解后,以氢氧化铵代替氢氧化钡中和水解产物,因而不需要用离心机除去硫酸钡沉淀。使用1-甲基咪唑作为催化剂,与用硫酸作催化剂于60℃处理1h相比较,乙酰化作用能在室温下在10~20min内完成,可以避免使用旋转式蒸发器。因为不需要高温蒸发,整个操作可在玻璃器皿中进行。一旦校正因子被得到,一次8个样品的分析能在8h内完成。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。