



热循环测试是测试电子封装可靠性的关键鉴定测试之一。封装的不同层之间热膨胀系数不匹配会引起较高的界面应力,在环境热负荷条件下这会威胁封装的可靠性。热循环测试是研究电子封装可靠性的一种标准加速测试方法。为了预测热循环过程中材料的性质,人们广泛采用有限元分析来模拟热循环载荷下的电子封装[34,35]。可靠性测试过程中电子封装发生失效的机理,可通过有限元分析进行研究,然而测试中的一些现象至今仍然未知。此外,封装中界面材料的特性与其他大块材料的特性十分不同,因此有必要在原子水平上研究界面材料的性质。
分子模拟技术被成功用来研究固化的环氧树脂和其他基底之间界面粘附的基本原理,并揭示了粘附形成及失效的分子机制[6-8]。Iwamoto[13,14]对微电子封装工业中粘附剂配方的影响进行了分子动力学模拟,并在电子封装应用领域提出了一些重要而有趣的研究结果[15,16]。然而,目前几乎无人研究热循环条件下材料的特性,特别是电子封装中广泛使用的EMC-Cu(环氧模塑料-铜)系统的性质。
Iwamoto[15]提出了一套分子动力学模拟程序来预测材料性能,研究了应力循环和过程分析,以得到材料可能的性能趋势,从而了解热循环下材料的失效机制。Fan等人[23]采用同样的分子动力学模拟程序研究了热循环测试中环氧模塑料(EMC)和氧化亚铜基底之间的粘附性。详细内容如下。
美国Material Studio公司的Accelrys软件被用来对热循环测试进行分子动力学模拟。该分子动力学模拟使用用于原子仿真研究的冷凝相优化分子电位(Con-densed-phase Optimized Molecular Potential for Atomistic Simulation Studies,COM-PASS)力场。COMPASS力场能够在不同条件下精确预测许多材料的材料特性。COMPASS力场也能精确预测由高分子、金属及其交界面所组成的系统的特性。
典型地,环氧模塑料内支配其粘附特性的核心化学结构是由环氧树脂和固化剂反应形成的。该研究模拟的环氧模塑料包括环氧树脂和固化剂。但该模拟不包括填料和颜料,因为填料和颜料需要大尺度的模型来模拟,而这超过了目前分子动力学模拟的范围。在这项研究中,EMC配方的基本成分是双酚A型缩水甘油醚(Digly-cidyl Ether of Bisphenol-A,DGEBA)环氧树脂和亚甲基联氨二苯胺(Methylene-diamine-dianiline,是一种MDA)固化剂。图3.1所示为DGEBA环氧树脂和MDA固化剂的固化反应过程。
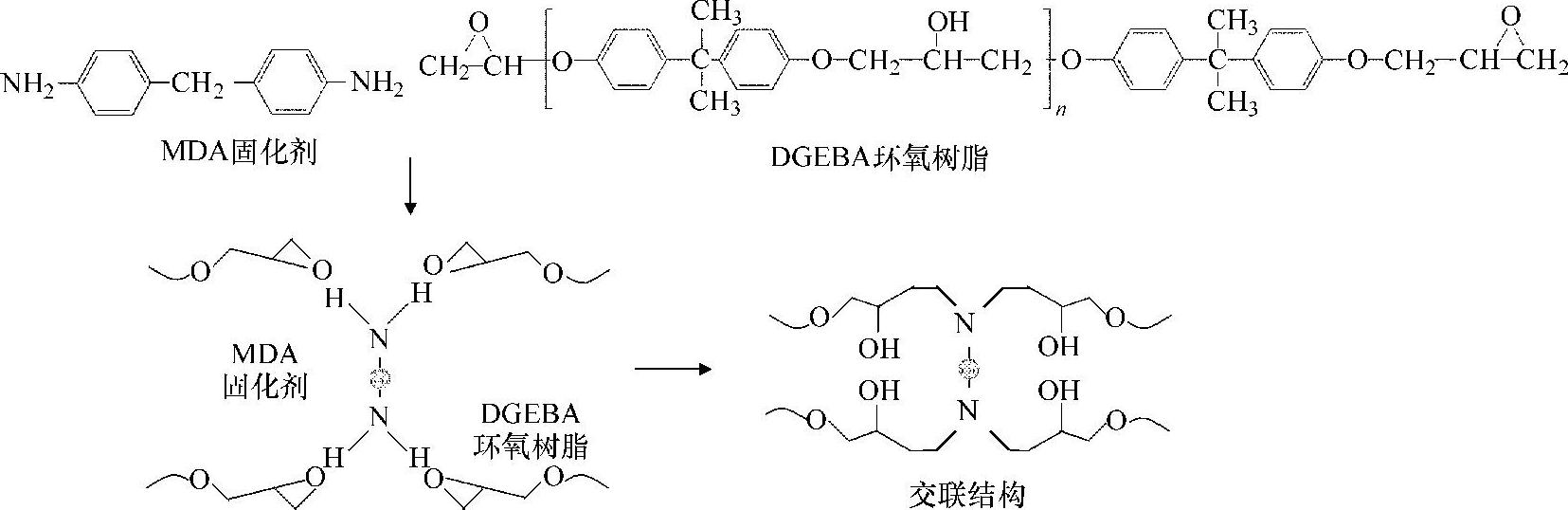
图3.1 环氧树脂和固化剂的固化反应过程
通过观察发现,在造型过程中,环氧模塑料/铜界面处的金属表面上发生了氧化反应,这对界面粘附性造成了很大影响。Cho等人[36]发现铜表面的化学成分从金属铜变成了氧化亚铜。Chung等人[37]进行了一项热循环测试,并发现所有剪切下的样品中只有氧化亚铜,而且氧化亚铜的含量随着热循环次数而变化。
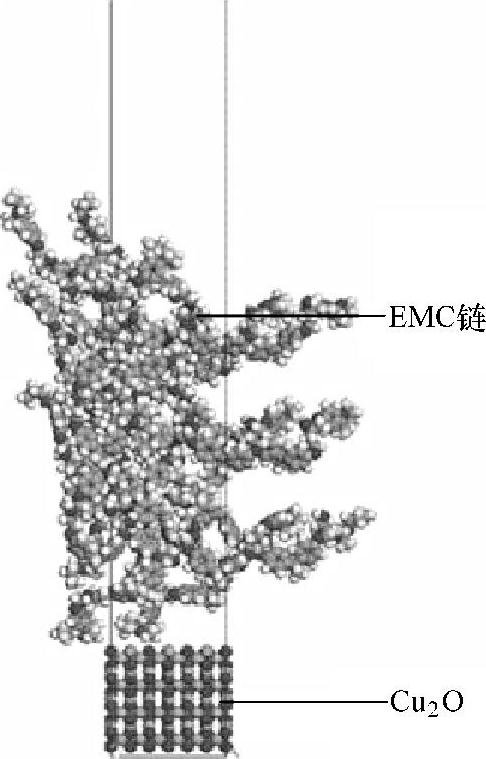
图3.2 环氧模塑料-氧化亚铜系统的分子模型
因此,在分子动力学模拟中只需考虑氧化亚铜的影响。为了研究铜基底上氧化亚铜的含量对环氧模塑料(EMC)与铜基底之间界面粘附性的影响,Fan等人[23]建立了两种分子动力学模型用于研究热循环测试。在第一种模型中,氧化亚铜原子的数量随着热循环的次数而变化,与Chung等人[37]进行的实验一样。而在第二种模型中,在整个热循环测试过程中,氧化亚铜的含量保持不变。这两种模型的矩形模拟网格均如图3.2所示。在x和y方向上矩形网格的大小为17.9×17.9Å2(1Å=0.1nm),而在垂直于环氧模塑料与氧化亚铜界面的平面上,矩形网格呈周期性分布。这两种模型使用的双材料系统包含EMC碎片和氧化亚铜原子。模拟单元中EMC环氧树脂链的顶部有很大的真空,以避免z方向上的EMC镜像间的相互作用。该模型单元的高度是100Å,氧化亚铜的厚度是20Å。所有氧化亚铜原子都是固定的,而所有的EMC链则可以自由移动。
根据Chung等人给出的实验条件[37],分子动力学模拟在175℃和4.3MPa下采用颗粒、压力和温度常数(Number of Particles、Pressure and Temperature,NPT)系综进行。随后整个结构运用颗粒、体积和温度常数(Number of Particles、Volume and Temperature,NVT)系综冷却至室温。
Chung等人[37]在标准JEDEC[1]温度曲线(JESD 22-A 104-B第M种情况)下进行了热循环测试。在这个研究中,环氧模塑料和Cu2O的热膨胀系数分别为45×10-6/℃和8×10-6/℃。利用环氧模塑料和氧化亚铜的热膨胀系数的不一致性,可用来帮助说明在一次热循环过程中加热和冷却步骤分别造成的变形。该分子动力学模拟为每个阶段(加热和冷却)采用不同的应变量,以便模拟一次热循环过程。该分子动力学模型采用了不同的应变量,以便能模拟如下两个过程:在加热阶段环氧模塑料挤向氧化亚铜基底;在冷却阶段环氧模塑料脱离氧化亚铜基底。整个系统在冷却温度和加热温度下放松。采用不同应变量重复上述的冷却和加热过程,从而完成整个热循环测试。(https://www.xing528.com)
图3.3所示为经过不同次数热循环后该系统的分子结构。结果发现,在热循环测试中环氧模塑料和氧化亚铜的界面处形成了很大的空隙。该空隙是因热循环测试过程中环氧模塑料和氧化亚铜的热膨胀系数不一致而引起的界面应力导致的。因此,在经过1800次热循环后,观察到环氧模塑料和氧化亚铜基底之间的粘附强度变小了。
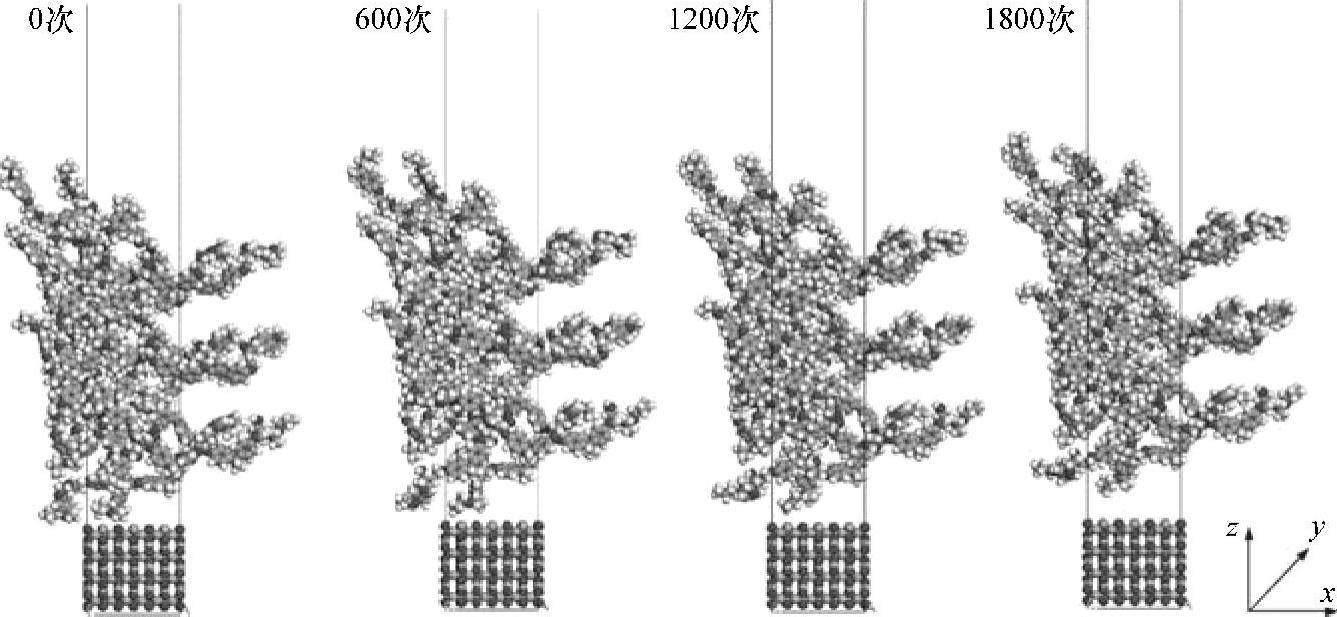
图3.3 不同次数的热应力循环后系统的分子动力学模拟快照
环氧模塑料和氧化亚铜基底的相互作用受静电力和范德华力的支配。可通过整个系统的总能量减去各个子系统能量的总和来获得界面结合能γ,如下所示[23]:
γ=ΔE/2A
ΔE=Etotal-(EEMC+ECu2O) (3.5)
式中,A为环氧模塑料与氧化亚铜基底之间的接触面积;Etotal为整个系统的总能量;EEMC为无氧化亚铜基底时环氧模塑料的能量;ECu2O为无环氧模塑料时氧化亚铜基底的能量。
Fan等人[23]计算了两个分子动力学模型中环氧模塑料与氧化亚铜基底之间的界面结合能,并以热应力循环次数为横轴,计算结果如图3.4所示。图中,对于第一种分子动力学模型,界面结合能先随热应力循环次数的增加而变大,而之后随着循环次数继续增加其界面结合能降低。对第二种分子动力学模型,界面结合能一般随着热循环的增加而减少。两种模拟之间的不同表明,环氧模塑料与氧化亚铜基底之间的界面粘附性受铜基底上氧化亚铜含量变化的支配,且铜基底上氧化亚铜的含量越高,环氧模塑料与铜基底之间的粘附强度就越大。
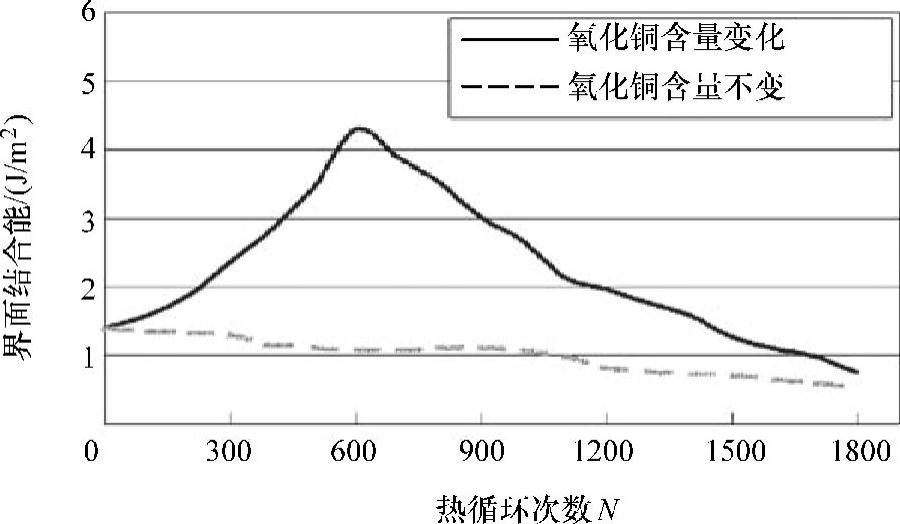
图3.4 界面结合能(是热循环次数的函数)
Chung等人[37]的实验结果证实了上述分子动力学模拟结果。一个类似的趋势表明,环氧模塑料与铜基底之间的界面粘附性受铜表面氧化物含量变化的支配。Su和Shemenski[38]也证明采用氧化亚铜能增加橡胶和铜线之间的粘附性。Ken-dall[39]也认为界面之间的粘附性受键合表面的特性,而非两种键合材料的整体性质支配。所有这些结论都与热循环测试的分子动力学模拟结果一致。现阶段,还不能采用定量方法将分子动力学模拟结果与实验测量进行比较评估,但是分子动力学模拟预测所得的定性趋势和实验观测结果相一致。
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。