



2.3.4 色谱法
色谱法所用的固定相和流动相是互不相溶的。固定相只能是液体和固体,流动相只能是液体和气体。当流动相为液体时称为液相色谱;当流动相为气体时则称为气相色谱。
色谱法根据其分离机理的不同进行分类。利用吸附剂表面对不同化合物的吸附性能差异达到分离目的,称为吸附色谱;利用不同化合物在流动相和固定相之间的分配系数不同而达到分离目的,称为分配色谱;利用分子大小不同、阻滞作用不同而实现分离的,称为排阻色谱;利用不同化合物对离子交换剂亲和力不同而进行分离的,称为离子交换色谱。此外,色谱法还根据进行分离的流动相和固定相之间极性的大小,分为正相色谱和反相色谱。
1.吸附色谱
吸附色谱(adsorption chromatography)的原理就是利用混合物中的各化合物对固定相吸附剂的吸附能力不同而达到分离的色谱方法。吸附色谱是在天然药物分离中应用最多的一种色谱方法,特别适用于脂溶性成分的分离,不适于蛋白质、多糖或者离子型亲水性化合物的分离。吸附色谱的分离效果取决于吸附剂的选择、洗脱剂的选择、待分离化合物的性质以及吸附色谱分离的操作过程。
(1)吸附剂的选择:吸附剂是吸附色谱在分离过程中使用的固定相。一般是多孔物质,具有很大的比表面积,表面上具有多孔吸附中心。吸附中心的多少和吸附能力的强弱直接影响吸附剂的性能。常用的吸附剂有硅胶、氧化铝、活性炭和硅藻土等。
1)硅胶:硅胶是吸附色谱最常用的吸附剂,约90%的分离工作都可以采用硅胶作为吸附剂。硅胶通常用SiO2・x H2 O表示,硅胶是由硅氧烷(siloxane)及—Si—O—Si—的交联结构组成,表面带有许多硅醇基的多孔性微粒。硅醇基是硅胶具有吸附能力的活性基团,它能与极性化合物或不饱和化合物形成氢键或发生其他形式的相互作用。被分离物质由于极性和不饱和程度不同,与硅醇基相互作用的程度不同而得到分离。
硅胶的吸附性能不仅与其含有硅醇基的数目有关,还与其含水量密切相关。水能与硅胶表面羟基结合成水合硅醇基而使其失去活性。因此随着水分的增加,硅胶的吸附能力降低。但将硅胶加热到100℃左右,水能可逆地被除去。若硅胶中含水量达12%以上,则吸附性能极弱,不能进行吸附色谱,只能进行分配色谱。若将硅胶在105~110℃加热30分钟,降低硅胶含水量,增强硅胶吸附力,这一过程称为活化。
硅胶是一种微酸性吸附剂,适于分离酸性和中性物质,同时硅胶表面的硅醇基能释放弱酸性的氢离子,当遇到较强的碱性化合物时,可通过交换离子分离碱性化合物。因此硅胶在酚类、醛类、生物碱、氨基酸、甾体及萜类等天然产物的分离过程中都有广泛的应用。硅胶的分离效率与其孔径、体积、粒度及表面积等几何结构有关。硅胶粒度较小,均匀性越好,分离效率越高;硅胶表面积越大,则与样品之间的相互作用越强,吸附力越强。硅胶的优点在于不像氧化铝有时与分离样品发生副反应,但硅胶的吸附性能和分离效率有时不如氧化铝好。
硅胶吸附色谱分离中,可选择的洗脱剂种类较多,为了达到好的分离效果,可以选择一定比例多种溶剂的混合物作为流动相。可以借助分析型硅胶薄层色谱摸索分离条件,实际色谱分离过程中经常采用从低极性溶剂逐渐递增极性的梯度洗脱方式进行。
2)氧化铝:氧化铝是最常用的吸附剂之一,是由氢氧化铝直接在高温下(400~600℃)脱水制得,因制备方法和处理方法的差异有碱性、中性和酸性三种:①碱性氧化铝(pH9~10),适用于一些碱性(如生物碱)和中性化合物的分离,由于有时碱性氧化铝能够与某些醛、酮、内酯类成分发生氧化、皂化、消除以及异构化反应,因此不适用于醛、酮、内酯类成分分离;②中性氧化铝(pH7.5),应用最多,适用于生物碱、挥发油、萜类、甾体以及在酸、碱中不稳定的苷类、内酯类化合物;③酸性氧化铝(pH4~5),适用于酸性物质如酸性色素、某些氨基酸等的分离。柱色谱氧化铝的粒度一般在100~160目之间,低于100目,分离效果差,高于160目,柱色谱流速太慢,导致分离样品易于扩散,分离效果降低。样品与氧化铝的用量一般在1∶(20~50)之间,色谱柱的内径与柱长比例在1∶(10~20)之间。柱色谱分离时,流速不宜过快,一般以每30~60分钟内流出液体的体积等同于所用吸附剂的量为宜。
3)活性炭:活性炭是使用较多的非极性吸附剂,主要用于分离水溶性成分。其特点是具有非极性的表面,能够吸附疏水性和亲脂性有机物质,吸附容量大,耐酸耐碱,化学稳定性好。色谱用活性炭主要有粉末状活性炭、颗粒状活性炭和锦纶活性炭,其具体特点见表2-5。
表2-5 色谱用活性炭的类型和特点
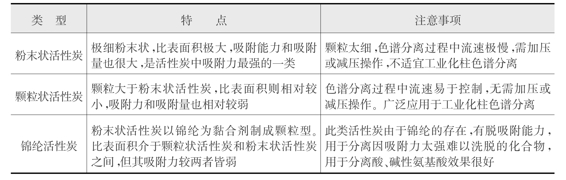
活性炭主要用于分离水溶性物质如氨基酸、糖类及某些苷类。活性炭的吸附作用与所选择的溶剂极性密切相关,在极性强的水溶液中吸附能力最强,洗脱能力最弱,在有机溶剂中吸附能力较弱,洗脱能力就增强。如以乙醇-水为例进行洗脱时,随着乙醇浓度的递增而洗脱力增加,有时亦用稀甲醇、稀丙酮、稀醋酸溶液洗脱。活性炭对极性基团多的化合物的吸附力强于极性基团少的化合物,对芳香族化合物的吸附力大于脂肪族化合物;对大分子化合物的吸附力大于小分子化合物。因此可以利用这些吸附性能的差别,将水溶性芳香族化合物与脂肪族化合物、氨基酸与肽、单糖与多糖分开。
使用前应先将活性炭于120℃加热4~5小时,使所吸附的气体除去。使用过的活性炭可用稀酸、稀碱交替处理,然后水洗,加热活化。由于活性炭的生产原料、制备方法及规格不统一,其对天然产物的吸附能力无法准确测定和控制,从而使其应用受到一定限制。
(2)待分离物质的性质:待分离的物质、固定相和洗脱剂共同构成了吸附色谱的三个要素。固定相即吸附剂的选择对于成功地进行色谱分离至关重要。选择适合的吸附剂作为固定相主要依据待分离对象的性质、分离的目的和吸附剂的性质进行决定。
对于被分离对象,考虑的主要因素有以下三方面:①化合物的极性:化合物的极性与分子结构及分子中所含官能团极性密切相关。一般来说,饱和碳氢化合物为非极性化合物,不被吸附剂吸附;随着化合物极性增加,吸附能力也增强,如分子中双键越多,吸附力越强,共轭双键延长,吸附力随之增加。值得注意的是,化合物结构中如果含有较多易于被吸附的基团,可能在硅胶或氧化铝上吸附得太牢而得不到分离。②化合物的酸碱性:硅胶由于含有硅醇羟基略带酸性,适用于分离微酸性和中性物质,而碱性物质易于被硅胶吸附发生相互作用,分离效果较差。而氧化铝略带碱性,适用于碱性和中性物质的分离,酸性物质因能与其发生反应,难以得到较好的分离效果,可以选用中性或酸性氧化铝进行分离。③化合物的溶解性:绝大部分的有机化合物都可以采用硅胶和氧化铝作为固定相进行分离,水溶性样品如糖苷类化合物、氨基酸类化合物等可以采用活性炭来分离。
(3)吸附色谱洗脱剂的选择:吸附色谱分离过程中洗脱剂的选择对组分分离效果影响极大。色谱分离过程实际上是组分分子与洗脱剂分子竞争占据吸附剂表面活性中心的过程,选择洗脱剂应同时考虑被分离物质的性质、吸附剂的活性及洗脱溶液的极性三个因素。分离强极性组分时,要选用吸附活性弱的吸附剂,以强极性洗脱剂洗脱。分离弱极性组分时,要选用吸附活性强的吸附剂,以弱极性洗脱剂洗脱。常用的单一溶剂洗脱剂的极性顺序为:石油醚<环己烷<二硫化碳<四氯化碳<三氯乙烷<苯<甲苯<二氯甲烷<三氯甲烷<乙醚<乙酸乙酯<丙酮<正丙醇<乙醇<甲醇<吡啶<水。洗脱剂由单一或混合溶剂构成:①以单一溶剂为洗脱剂时,溶剂组成简单,分离重现性好,但其极性固定不变,因而洗脱能力有限,分离效果不佳;②实际操作中常采用二元、三元甚至多元溶剂组分,有时为了提高分离度,在洗脱剂中还需加入少许酸、碱,促使某些极性物质的斑点集中。
(4)吸附色谱操作技术:吸附色谱按其操作方式可分为薄层色谱和柱色谱。薄层色谱适用于分析或少量样品的分离制备;经典的柱色谱法由于样品容量大,适用于天然产物的制备分离。
1)吸附薄层色谱:吸附薄层色谱是一种简便、快速、微量的色谱方法,其分离原理与柱色谱基本相似,故常作为选择色谱分离条件的参考。薄层色谱就是将超细吸附剂涂布到玻璃片或铝箔等平面载体上,形成固定相,将样品点样于薄层上,放置在一个用展开剂饱和的密闭容器内,展开剂依靠固定相的毛细作用和固定相做相对移动,使物质发生分离。
比移值(R f)是薄层色谱中用于表示物质移动的相对距离。

R f值随分离化合物的结构、固定相、流动相的性质、温度等多种因素的不同而变化。当各种因素固定时,R f就是一个特定的常数,因此可以作为物质定性鉴别的依据。但有时由于影响因素较多,实验数据会出现和文献报道数据不完全一致的情况,因此在鉴别时应采用与对照品同步展开分析。
薄层色谱在天然药物中常用于柱色谱分离的条件摸索、药材成分的定性鉴定、分离化合物的纯度检验以及分析提取物中化合物的组成、有效(或主要)成分的鉴别等。鉴定药材主要成分时,将药材提取液、对照药材提取液和单体对照品同时点样于同一薄层板上,色谱展开后,若样品液与对照药材提取液在同一位置上显相同颜色斑点,说明该药材与对照药材为同一药材。当然,两者应与单体对照品相对应。鉴别化合物的真伪就是将化合物和标准品分别点在同一块薄层上,用选定溶剂展开后,观察两者在薄层色谱中的R f值。若用三个不同展开溶剂进行色谱分析,两者R f都相等,初步推测化合物和标准品为同一物质。若鉴别化合物纯度时,采用三种不同溶剂系统进行薄层分析,结果在三个薄层色谱板上都为单一斑点者,可以推测该化合物为单一化合物。此外在薄层色谱中可以采用特异性显色剂对展开完成的薄层板进行显色反应,不仅可以确定植物中可能存在的化合物种类,也可以对色谱行为中R f值相同的化合物与对照品进行有效鉴定。
在确定药材进行柱色谱分离前,往往需要借助薄层色谱确定吸附剂和洗脱剂的选择;在柱色谱分离过程中,则需要利用薄层色谱分析洗脱下来的组分中化合物的组成和相对含量。通过薄层色谱摸索得到比较满意的分离条件,即可将此条件用于柱色谱。用薄层色谱进行某一组分的分离摸索条件,若两个样品的R f值之差大于0.2时,可达到分离目的。同样的溶剂系统条件下,经柱色谱能得到较好的分离。
此外,薄层色谱法在天然药物品种、药材及其制剂真伪的检查、质量控制和资源调查,对控制化学反应的进程,反应副产品产物的检查,中间体分析,化学药品及制剂杂质的检查,临床和生化检验以及毒物分析等领域也都得到了广泛的应用。
2)吸附柱色谱:目前最常用的色谱类型是各种柱色谱,经典的柱色谱是将作为固定相的吸附剂装在色谱柱中,从上端加入洗脱剂,使洗脱剂由上往下洗脱,使各种成分根据其在吸附剂上的吸附效果不同而依次流出色谱柱。在洗脱时,可在柱的下端依次收集洗脱液并进行检查。柱色谱的基本装置示意图如图2-11所示。
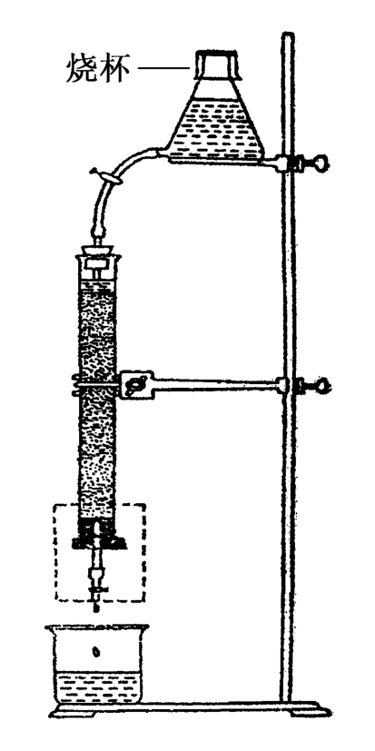
图2-11 柱色谱基本装置示意图
柱色谱的基本操作包括以下步骤:
①装柱:装柱质量的好坏,是柱色谱法能否成功分离纯化物质的关键之一。一般分为干法装柱和湿法装柱两种。干法装柱就是将吸附剂通过漏斗慢慢加入到柱中,吸附剂一定要压实,或者上端小心用平物将其压紧,操作时用力均匀。吸附剂一定要填装松紧一致。吸附剂填充完毕后,打开下端活塞,由上端缓慢加入初始洗脱剂,除尽柱子中的气泡,平衡色谱柱。湿法装柱则是取适量吸附剂与洗脱用的初始溶剂充分混合、搅拌或浆糊状,使其具有一定的流动性,除去气泡,沿色谱柱内壁徐徐倒入柱中,同时打开下端活塞,让洗脱液呈滴流出。填装要均匀,不能分层,柱子中也不能有气泡。还要注意在装柱和洗脱过程中不能让洗脱剂流完、干柱,应在吸附剂填料的柱平面上端始终保有一定液面。
②加样:加样量的多少直接影响分离的效果。一般来讲,加样量少些,分离效果比较好。通常加样量应少于20%的操作容量,体积应低于5%的柱床体积。最大加样量必须在具体条件下多次试验后才能决定。
加样的方式也可以选用干法和湿法两种加样方式。干法加样在待分离样品在初始洗脱溶剂中溶解不好时采用,选用溶解性好、易挥发的溶剂将样品溶解,按照样品和吸附剂的比例为1∶(2~3),根据样品的溶剂的体积,可以一次性或分次均匀加入到吸附剂中,挥干溶剂后,将吸附有样品的吸附剂均匀添加到色谱柱的吸附剂上面。干法拌样时切勿因样品溶解体积过大而一次性加入吸附剂,导致样品未被吸附剂吸附而在溶剂挥干过程中重新形成固体,混在吸附剂中直接进行加样,将影响后续的色谱分离工作。湿法加样时应缓慢小心地将样品溶液加到固定相表面,尽量避免冲击柱床表面,以保持基质表面平坦。如果样品能够溶解在初始的洗脱液中,则溶解后小心滴加到色谱柱的吸附剂表面,动作轻,不要扰动柱床表面。要求样品溶液体积尽量小,浓度高,才能够形成谱带狭窄的原始带,利于分离。(https://www.xing528.com)
③洗脱:选定洗脱液后,洗脱的方式可分为简单洗脱和梯度洗脱两种。简单洗脱就是采用单一溶剂进行洗脱,直至色谱分离过程结束。如果不能得到满意的分离效果则可以采用梯度洗脱或者分步洗脱。梯度洗脱就是通过增加洗脱剂的极性,逐渐提高洗脱能力,让各组分得到更好的分离效果。分离过程中注意洗脱剂的极性应该缓慢增大,才有利于混合物的分离。
④收集、鉴定:收集的组分进行薄层色谱鉴别,根据鉴别结果进行组分合并。
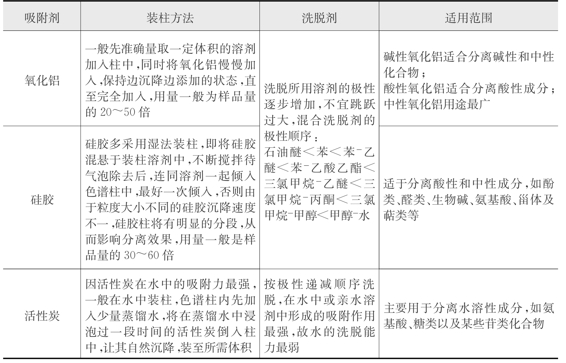
吸附柱色谱的具体操作方法见表2-6所示。
表2-6 吸附色谱常用固定相的选择及使用方法
2.分配色谱
分配色谱是基于混合物各组分在固定相与流动相之间的分配系数不同而实施分离的一种色谱方法。
(1)基本原理:分配色谱中将作为固定相的溶剂牢固吸附或以化学键形式结合在惰性固体物质表面,这些惰性固体物质起到支持固定相溶剂的作用称为支持剂。分配色谱分离原理就是不同的化合物在互不相溶的两相溶剂中分配系数不同,导致色谱过程中迁移速度各异,分配系数小的溶质在流动相中分配的数量多,移动快,分配系数大的溶质在固定相中分配的数量多,移动慢,因此可彼此分开。
(2)分配色谱的分类:①按照支持剂的不同可分为:纸色谱、硅胶分配色谱等;②按照支持剂的装填方式可分为:柱色谱、薄层色谱;③按照流动相的状态可分为:液-液分配色谱、气-液分配色谱;④按照流动相和固定相极性的大小分为:正相分配色谱和反相分配色谱。
(3)纸色谱:以滤纸作为支持物的分配色谱。固定相为滤纸上吸附的水,滤纸是支持剂,流动相由有机溶剂和水组成。滤纸纤维与水有较强的亲和力,能够吸取22%左右的水,其中6%~7%的水是以氢键的形式与纤维素的羟基结合。将样品点样于滤纸上,当流动相(展开剂)在滤纸上展开时,样品就在流动相和水相之间进行反复分配,最终由于各组分在两相中分配系数不同,迁移速度不同,从而使样品在滤纸上实现分离。纸色谱的展开剂常由有机溶剂和水组成,如常用的有水饱和正丁醇或正丁醇-醋酸-水(4∶1∶5)。展开剂的选择应在参考文献的基础上,对于待分离组分在该系统中有很好的溶解能力,不与样品发生化学反应,且各组分在该溶剂系统中的R f值差异也较大。
色谱展开过程中,当样品中所含溶质较多、样品之间的R f值比较接近,单向色谱过程不易明显分离时,可采用双向纸色谱法。该法是点样后将滤纸在某种展开剂中按一个方向展开以后即予以干燥,再转向90°,在另一种展开剂中展开,待溶剂到达所要求的距离后取出滤纸,干燥显色,从而获得双向色谱。应用这种方法,如果溶质在第一种溶剂中不能完全分开,而经过第二种溶剂的色谱能得以完全分开,大大地提高了分离的效果。纸色谱还可以与区带电泳法结合,能获得更有效的分离方法,这种方法称为指纹谱法。
在纸色谱过程中特别需要注意的是点好样的滤纸放入层析缸中,先不要浸入展开剂,使滤纸及层析缸中空气被溶剂蒸气饱和后,再使滤纸进入展开剂开始展开,分离效果更好。
(4)反相色谱技术
1)正相色谱和反相色谱的定义:通常把固定相极性大于流动相极性,化合物流出色谱柱的极性顺序是从小到大的色谱过程称为正相色谱。将固定相极性小于流动相极性,化合物流出色谱柱的极性顺序是从大到小的色谱过程称为反相色谱。
2)反相色谱的固定相:反相色谱分离中固定相吸附剂是由键合相硅胶构成的。利用硅胶表面的硅醇基能够与各种不同极性基团形成化学键合相,如其与氰乙醇、正辛醇以及聚乙二醇等在一定温度下加热脱水生成单分子键合固定相(Si—O—C型),与十八烷基三氯硅烷生成烷基化学键合相(Si—O—Si—C型),还可以利用化学试剂如SOCl2将硅胶表面氯化后,再与各种有机胺反应生成具有Si-O-Si-N键的各种不同极性基团的化学键合相。这些键合相针对不同结构类型的天然产物具有不同吸附选择性,从而达到分离目的。键合相硅胶在高效液相色谱中应用最多,常用的是带有十八烷基主链的硅胶填料,一般称为ODS或C18硅胶,目前已经商品化的反相填料还有C4、C8、苯基、氨基、氰基等。这些固定相具有可以反复使用,不可逆吸附少,待分离样品破坏少、损失少等优点。
3)反相色谱的使用:利用键合相硅胶进行反相色谱分离时,流动相大多选择甲醇-水、乙腈-水、乙醇-水等组合作为洗脱剂。
当待分离组分吸附到固定相上之后,通过改变流动相的极性来改变待分离组分与固定相之间的作用,达到洗脱的目的。随着流动相极性的减小,其洗脱能力逐渐增强,具体实践中可以通过反相薄层色谱和分析型高效液相色谱摸索分离制备的条件。
【思考题】
1.药材原料的内部构造、主要化学组成与天然药物提取工艺之间的关系是什么?
2.以植物的不同器官部位作为原料设计提取工艺时应注意什么?
3.药材原料在提取之前常规破壁的方法有哪些?
4.天然药物的传统提取分离方法有哪些?基本原理是什么?适用于哪些成分的分离?
5.有哪些常用的吸附剂?在分离上有哪些区别?
6.正相色谱和反相色谱的区别是什么?
7.任选一类天然药物,采用传统的提取分离方法设计一个提取分离工艺流程。
【参考资料】
[1]郑汉臣,蔡少青.药用植物与生药学(第4版)[M].北京:人民卫生出版社,2003:9-94
[2]郑俊华.生药学(第3版)[M].北京:人民卫生出版社,2003:139-325
[3]徐怀德.天然药物提取工艺学[M].北京:中国轻工业出版社,2008:22-68,151-178
[4]吴立军.天然药物化学(第5版)[M].北京:人民卫生出版社,2007:18-35
[5]卢晓江.中药提取工艺与设备[M].北京:化学工业出版社,2004:37-55
[6]卢艳花.中药有效成分提取分离技术[M].北京:化学工业出版社,2005:2-8,12-20,24-42
[7]徐任生,叶阳,赵维民.天然药物化学导论[M].北京:科学出版社,2006:4-24
[8]吴立军.实用天然有机产物化学[M].北京:人民卫生出版社,2007:6-38
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。